Research
* microbial ecology and evolution in marine oxygen minimum zones * microbial genomics and metagenomics * microbial symbiosis

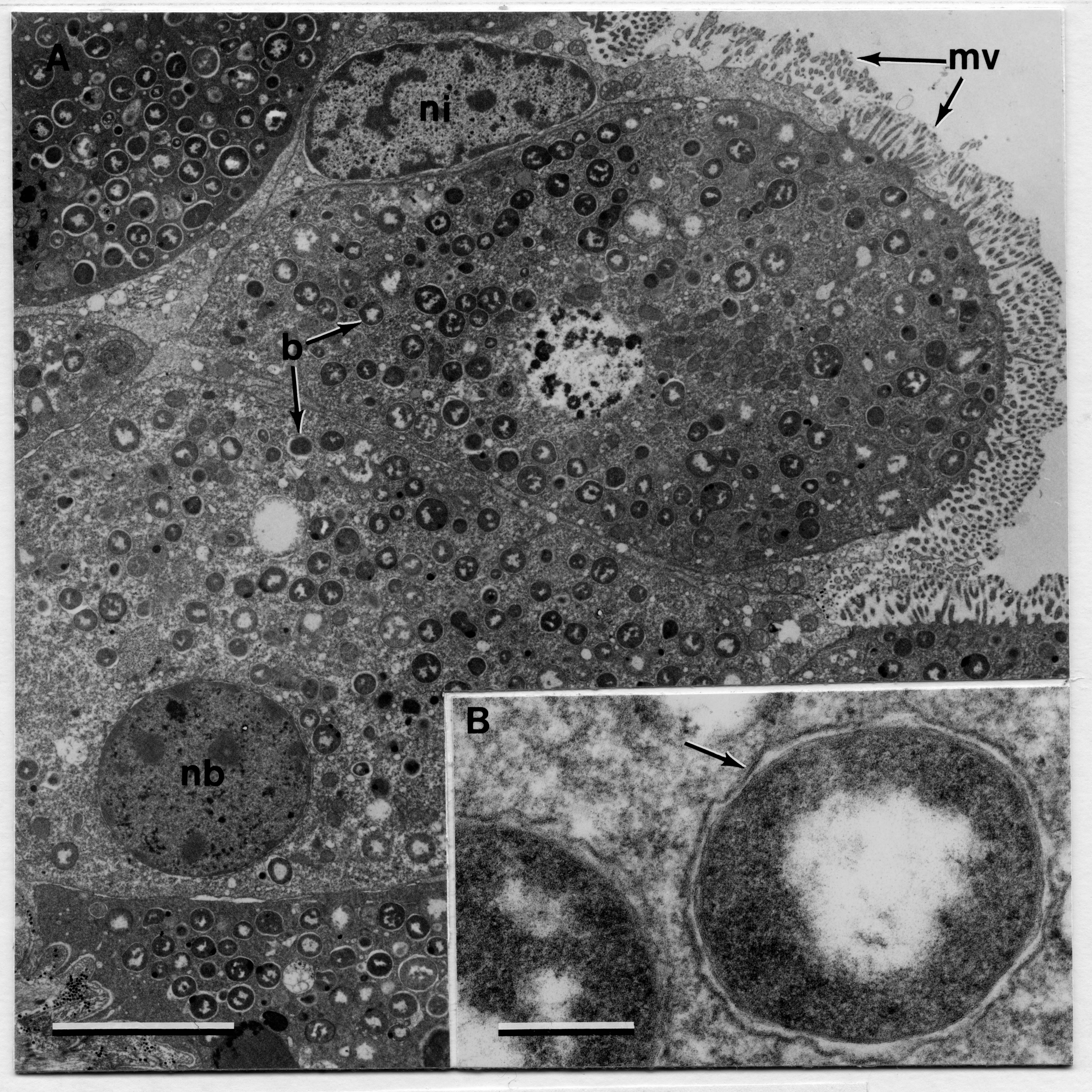
2500 m depth.
Bacteria and Archaea constitute the overwhelming majority of genetic and metabolic diversity on this planet. To understand these organisms in their native habitats, environmental microbiologists are tasked with two fundamental questions. First, how do ecological and evolutionary processes (e.g., environmental gradients, symbiosis, competition, recombination, natural selection) create and structure genetic diversity? Second, how is this genetic diversity linked to the diverse biogeochemical functions of microorganisms in nature?
Our research explores these questions for marine microorganisms, using the tools of genomics and molecular biology. We are particularly interested in how marine microbial diversity, genome evolution, and physiology are structured by environmental gradients, notably dissolved oxygen concentration, as well as by interactions with other organisms (through symbioses for example).
A primary focus of the lab is to understand microbial communities in marine oxygen minimum zones (OMZs) - see below. These microbially-dominated ecosystems support functionally diverse but under-characterized commmunities of bacteria, archaea, and microbial eukaryotes whose metabolisms play critical roles in global biogeochemical cycles. We also study marine symbioses between bacteria and other organisms, including marine invertebrates. Some symbiotic bacteria are closely related to major clades of free-living OMZ bacteria. These groups are therefore natural systems for exploring the role of symbiosis in structuring bacterial genome content and ecology.
Our research integrates the broad fields of marine biology, microbiology, and molecular evolution and genomics. This work has both descriptive and experimental components, and involves a blend of field, molecular, and bioinformatic techniques, the latter focused (primarily) on the analysis of high-throughput sequence datasets. We welcome inquiries from potential students, post-docs, and collaborators who share these interests.


Eastern Tropical North Pacific, summer 2013
Oxygen minimum zones
A major challenge for earth scientists is to understand how oceans respond to decreasing oxygen levels. Areas of low oxygen, oxygen minimum zones (OMZs), are predicted to increase in both expanse and frequency in response to climate warming and human modifications of coastal zones. Such shifts have profound consequences for biogeochemical cycles, as OMZs are critical sites for microbially-mediated transformations of carbon, nitrogen, and sulfur, including releases of key greenhouse gases (e.g., N2O). OMZs occur throughout the world's ocean but vary substantially in biological, chemical, and physical properties, notably the levels of primary production and oxygen availability (for a review, see Ulloa et al. 2012, PNAS, 109: 15996-16003). How this variation differentially structures microbial taxonomic composition, population-level genomic subdivision, and metabolic processes is poorly understood. This is due in part to a lack of genomic and metagenomic (community DNA) data enabling comparisons across diverse low-oxygen systems.
Our research uses genomics, meta-omics (see below), and physiological measurements to explore microbial diversity and metabolism across a range of low-oxygen systems, with a particular emphasis on OMZ bacteria that use inorganic sulfur compounds for energy metabolism. Specifically, we study pelagic microbial communities in two prominent end-members in the spectrum of OMZ types, the permanent anoxic OMZs (AMZs) of the Eastern Tropical Pacific Ocean where O2 can fall below detection (< 15 nM), and the seasonally hypoxic “deadzone” in the Gulf of Mexico (GoM; O2 < 20 uM).
Research at these sites is funded by generous grants from the US National Science Foundation (award 1151698) and the Alfred P. Sloan Foundation, and has thus far involved cruises to both the GoM (2012) and the Eastern Tropical North Pacific (ETNP) AMZ south of Baja California (2013), with additional cruises scheduled for 2014 and 2015. These field campaigns have involved participants at all levels of scientific training (undergraduate to senior PI), collaborations from multiple US and international institutions, and cross-disciplinary sampling and analytical approaches. Our work at these sites, notably the ETNP AMZ, is designed to complement existing research efforts at these and at other oceanographically diverse OMZ sites. A broad goal of this research is to facilitate interactions among diverse communities (oceanographers, microbial scientists, geochemists) to develop an integrated understanding of marine ecosystem response to oxygen depletion and climate change.
Community genomics and gene expression - Metagenomics and Metatranscriptomics
A holistic understanding of microbial diversity and function requires studies targeting varying levels of biological complexity, from individual cells to entire communities and ecosystems. Advances in DNA sequencing have transformed our capacity to study microorganisms across complexity gradients. Entire genomes can now be sequenced within hours from DNA originating from single cells, facilitating comparative studies of genome architecture and content. Additionally, high-throughput shotgun sequencing can characterize the diverse pool of genes (DNA) and expressed transcripts (RNA) of an entire microbial community (the metagenome and metatranscriptome, respectively).
In combination with single-genome or metagenome analyses, we study microbial community gene expression using high throughout methods (e.g., Illumina technology) to sequence the metatranscriptome of microbial communities [for examples of these methods, see Frias-Lopez et al. 2008, PNAS, 105:3805-3810; Stewart et al. 2010, ISME J, 4:896-907]. The resulting datasets contain hundreds of thousands to millions of sequence fragments (reads) from both the transcriptionally active gene pool and the pool of non-coding RNA molecules (e.g., ribosomal RNA, small RNAs). When coupled to metagenomic data, metatranscriptomic analyses help clarify the link between the genetic potential of a community and its actual functional state (inferred from the transcript pool), reveal novel molecular and ecological adaptations to the geochemical environment, and inform theory about the molecular evolution of highly expressed genes. This work has focused primarily on bacterioplankton communities, sampled either directly from the marine environment or manipulated within an experimental framework (e.g., via mesocosm or bioreactor treatments). However, we are also applying these methods to other systems, including microbial symbioses.
Symbiont Evolution and Physiology
Symbiosis, defined broadly as a long-term interaction between species, is among the most pervasive evolutionary and ecological strategies in marine ecosystems, impacting fundamental processes such as speciation, ecosystem structuring, primary production, nutrient cycling, and disease. Marine microbial symbioses span a striking diversity of hosts and symbionts from all three Domains of life. These associations vary widely in key biological factors, including the extent to which symbionts affect host fitness (e.g., mutualism vs. commensalism vs. parasitism), integrate into host tissue, and exchange genetic material with free-living microorganisms. Though such factors have been explored for several well-studied associations, such as coral-algae mutualisms, most marine symbioses remain understudied.
Our research explores the evolution and physiology of symbioses between bacteria and marine invertebrates. We are particularly interested in symbioses involving chemoatotrophic bacteria. These symbioses, in which bacteria use the energy of reduced chemicals (e.g., sulfide, methane) to fix carbon for their host, have been observed in seven host phyla, play vital ecological roles as primary producers and ecosystem engineers in reducing habitats (e.g., anoxic sediments, hydrothermal vents), and are potential models for the ancient prokaryote-eukaryote associations that gave rise to the eukaryotic cell. In addition, several of these symbionts are closely related to free-living bacteria that mediate carbon fixation and sulfur cycling in the OMZ water column (e.g., the sulfur-oxidizing SUP05 lineage).
Genomic and meta-omic methods can provide exciting insight into the molecular basis of these associations. Working with collaborators, we are currently analyzing the metagenomes of sulfur-oxidizing symbionts from diverse marine environments. We have also developed transcriptomic techniques for studying gene expression in intracellular chemoautotrophic symbionts, enabling a community-level analysis of how symbiont metabolism and cell function vary in response to environmental conditions, geography, or the developmental or physiological state of the host. Using these methods, we can explore a range of questions: How does symbiont genome evolution and physiology vary in response to symbiont transmission mode and the level of symbiont-host specificity (e.g., free-living versus host-associated lineages)? How are symbiont and host genome diversity structured within and across populations and in response to factors external to the symbiosis, such as the genetic composition of a free-living symbiont community? Answering these questions for functionally diverse associations sheds important light on the role of microbial symbiosis as a driver of biological innovation.